Medical Devices Regulation (EU) 2017/745 - MDR

As a trusted notified body, DNV can help you navigate the complexity of the EU’s Medical Device Regulation and get access to the EU market.
On 5 May 2017, two new regulations on medical devices were published and came into force on 25 May 2017:
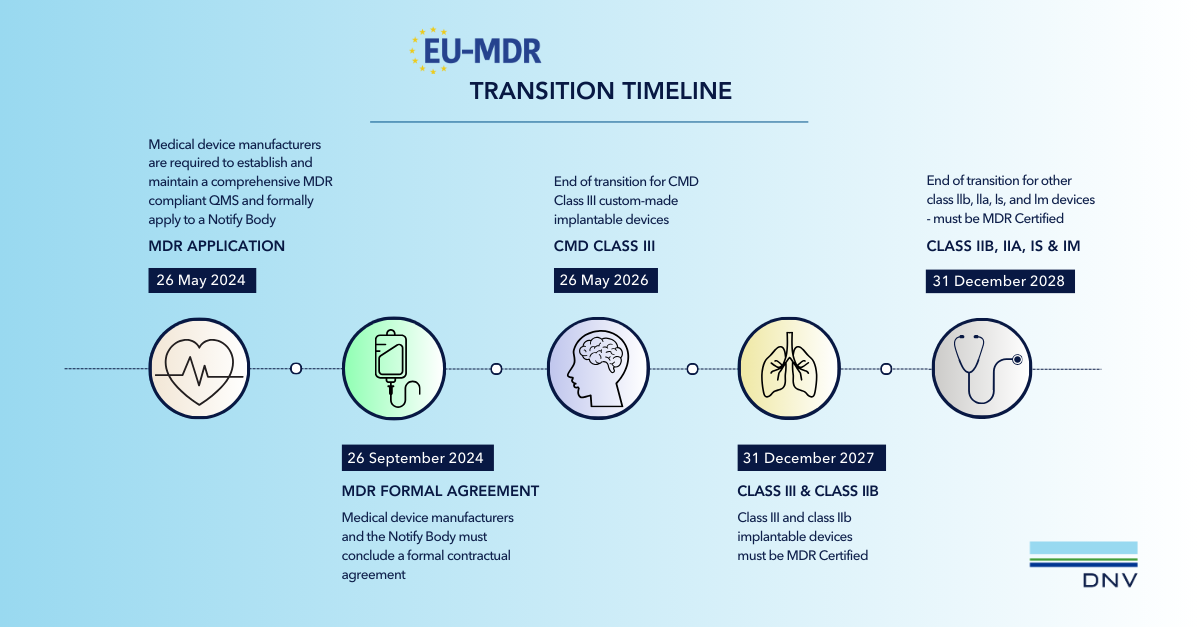
- The Medical Devices Regulation (MDR) became mandatory on 26 May 2021, requiring all medical devices placed on the European market to comply unless they fall under specific transition arrangements outlined in the regulation’s amendments. The transitional requirements are detailed below.
- The In Vitro Diagnostic Medical Devices Regulation (IVDR) has been in effect since 26 May 2022. However, like the MDR, transitional arrangements allow certain devices to continue being placed on the market under specific conditions.
- For the UK market, European CE marking is currently accepted until specified deadlines, which vary depending on device type (IVDs, general medical devices, or active implantable devices) and risk classification. The relevant requirements are outlined below.
What is Medical Devices Regulation (EU) 2017/745 - MDR
Medical Devices Regulation (EU) 2017/745 has replaced the Medical Device Directive (MDD) and the Active Implantable Medical Device Directive (AIMD), whereas the IVDR has replaced the In vitro Diagnostic Directive (IVDD). Both regulations bring a series of important improvements to conformity assessment for medical devices with the intention to:
- Improve the quality, safety and reliability of medical devices placed on the European market.
- Strengthen transparency of information related to medical devices for consumers and practitioners.
- Enhance vigilance and market surveillance of devices in use.
The extent to which these changes will affect your operations will depend on the type of device manufactured and the role you hold (manufacturer, importer, authorized representative). It is however anticipated that in most cases significant changes must be made to compliance process, quality management system, and technical documentation and successfully implemented, and certified as relevant to device classes and roles of your organization, before compliance with the regulations can be achieved.
Refer to the document on the application and conformity assessment process for medical devices for more details on the procedure.
Continued validity of MDD certificates
MDD certificates remain valid until the dates specified in the amendment to Regulation (EU) 2023/607, provided that the manufacturer and the notified body have agreed on continued surveillance of the certificate and that no significant changes have been made to the device’s design or intended purpose. The regulation, along with various guidance documents and a short factsheet, can be found on the European Commission's homepage.
Migration of MDD certified devices to MDR
The changes to the MDR and IVDR are often seen as a revision of the requirements. In reality, it is a new regulation and should be treated as such. In general, the concepts and the requirements of the MDD and IVDD remain part of the new legislation but significant additional requirements have been added and changes made. There are a number of new requirements that manufacturers need to address before an application for certification to the MDR and IVDR can be made.
A new application for certification must be made by the legal manufacturer which will commence a new certification cycle, with validity up to 5 years. Manufacturers shall successfully complete a Notified Body review of technical files and an initial on-site audit activity to verify implementation before certification will be granted. The review of technical files is based on sampling; however this is not permitted for Class IIb implantable or Class III devices, as all such devices must undergo review. Manufacturers shall ensure their technical files and design dossiers are MDR compliant prior to application.
MDR Certification Process
In order to support your migration to MDR, we have prepared a description of the application and conformity assessment process for medical devices, which calls out the activities needed to obtain and maintain certification. Note: the document is not exhaustive as to the activities performed by the notified body in relation to the manufacturer. The full list is part of the certification agreement between the Notified body and the Manufacturer.
Getting ready for certification
Whether migrating from MDD to MDR or just starting out, it is essential to first understand the regulation and requirements. As mentioned, there are substantial changes needed for the quality management system and technical documentation when compared to the Medical Device directive.
As a notified body, DNV Product Assurance cannot guide you on achieving compliance, as this would compromise our impartiality. DNV offers training to help you understand the requirements of the regulation and relevant standards (region-dependent). We conduct the certification process transparently, clearly outlining the necessary steps to achieve certification in accordance with regulations and market standards such as ISO 13485 (medical device quality management system).
UKCA specific requirements
The UK government has introduced measures allowing CE-marked medical devices to be placed on the Great Britain market according to the following timelines:
- General medical devices that comply with the EU Medical Devices Directive (EU MDD) or the EU Active Implantable Medical Devices Directive (AIMDD), with a valid declaration and CE marking, can be placed on the Great Britain market until either the certificate expires or 30 June 2028, whichever comes first.
- In vitro diagnostic medical devices (IVDs) that comply with the EU In Vitro Diagnostic Medical Devices Directive (IVDD) can be placed on the Great Britain market until either the certificate expires or 30 June 2030, whichever is earlier.
- General medical devices, including custom-made devices compliant with the EU Medical Devices Regulation (EU MDR), and IVDs compliant with the EU In Vitro Diagnostic Medical Devices Regulation (EU IVDR), can be placed on the Great Britain market until 30 June 2030.
- These timelines do not apply to Class I medical devices and general IVDs under the EU directives if their conformity assessment under the relevant directive and the EU MDR or EU IVDR does not involve a notified body. Likewise, they do not apply to custom-made devices compliant with the EU MDD or EU AIMDD.
Self-declared CE-marked Class I medical devices can continue to be placed on the Great Britain market beyond 30 June 2023 if they meet the following criteria:
- They are self-declared in compliance with EU MDR requirements (until 30 June 2030), or
- They were self-declared under MDD requirements before 26 May 2021, provided that notified body involvement was not required under MDD but is required under EU MDR (until 30 June 2028). This includes upclassified devices and reusable surgical instruments.
Class I medical devices with a sterile or measuring function that hold a valid MDD certificate can be placed on the GB market until 30 June 2028.
All devices must be registered with the MHRA before being placed on the GB market. In Great Britain (England, Wales, and Scotland), devices must comply with the UK MDR 2002, with transitional provisions allowing CE-marked medical devices to be accepted as outlined above for registration with the MHRA.
DNV designation
DNV is designated by the Norwegian Health Authorities and the European Commission as a Notified Body 2460 for Medical Devices Regulation (EU) 2017/745 (MDR). The designation is granted for all technology types applied for, including the highest risk Class III devices, ensuring continuity of European market access for our customers’ life-saving medical technology.
This consolidates the two previous MDD notifications for DNV Presafe and Presafe Denmark AS. The reason is to ensure we can provide a more effective service for our customers by combining our global technical resources (over 140 specialists in over 20 countries) under one Notified Body system.
DNV has submitted its application for In-vitro Diagnostic Devices Regulation (EU) 2017/746 (IVDR) to provide a global service for this rapidly growing sector. The application process is in its final stages.
DNV is in the process of becoming an Approved Body for issuing UKCA certificates and is expected to receive designation as a UK Approved Body later this year.



